






 In my early Thinking days, witnessing multiple neurological symptoms appear and then fade, I was introduced to the complexity of mitochondrial diseases or “mito” for short. I didn’t know anything about mitochondria or metabolism. No recollection from middle school science. Never heard mention at the pediatrician’s office. Not until our first series of appointments with a second neurologist was this topic casually introduced – through lab results and subsequent conversation of “ruled out” diagnoses. The vastness of metabolics reminded me of a scene from Willy Wonka and the Chocolate Factory where Gene Wilder opens the factory door to “Pure Imagination.” My reactions? Astonishment. Awe. Disbelief. I was giddy with possibilities.
In my early Thinking days, witnessing multiple neurological symptoms appear and then fade, I was introduced to the complexity of mitochondrial diseases or “mito” for short. I didn’t know anything about mitochondria or metabolism. No recollection from middle school science. Never heard mention at the pediatrician’s office. Not until our first series of appointments with a second neurologist was this topic casually introduced – through lab results and subsequent conversation of “ruled out” diagnoses. The vastness of metabolics reminded me of a scene from Willy Wonka and the Chocolate Factory where Gene Wilder opens the factory door to “Pure Imagination.” My reactions? Astonishment. Awe. Disbelief. I was giddy with possibilities.
With this year being our personal “10th year of Mito Awareness,” I have an odd sense of reflection this year. That week of testing and evaluations was eye-opening and empowering. I truly thought we would soon have a firm genetic diagnosis and a clear road map of our kids’ health. But that is not where we are ten years later. The kids are older with a few more non-invasive supports in place, and science has caught up to an intersection we passed by years ago called “Label my child’s symptoms.” But in those 10 years what exactly has changed within the mito community in research of research and treatment approach?
Mitochondria basics of diagnosis:
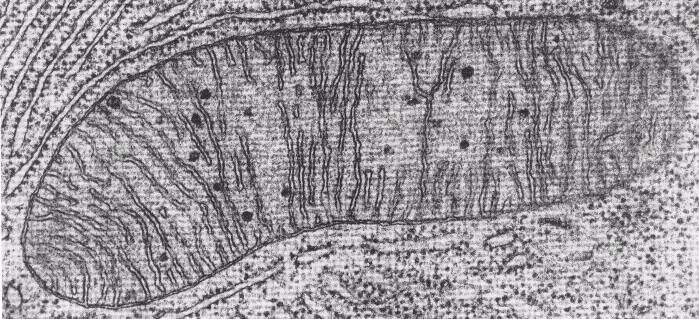 Alluding to the hundreds of processes it takes to be a human, we use the expression “eat to live.” We eat so we can keep our bodies functioning: our hearts beating, muscles moving, brain thinking. Our lungs breathe in oxygen which travels through the bloodstream to body tissues and into the mitochondria. Mitochondria use that oxygen and energy from the breakdown of the food we eat to make a chemical called ATP (adenosine tryphosphate). Without oxygen or nutrition, we can’t thrive. So we can’t just eat to live, we have to breathe as well. Breathing is the difference between being alive and dead — in more ways than one! But of course, the lungs can’t do their work unless they are already supplied with energy, which poses a “which came first: the chicken or the egg?” situation.
Alluding to the hundreds of processes it takes to be a human, we use the expression “eat to live.” We eat so we can keep our bodies functioning: our hearts beating, muscles moving, brain thinking. Our lungs breathe in oxygen which travels through the bloodstream to body tissues and into the mitochondria. Mitochondria use that oxygen and energy from the breakdown of the food we eat to make a chemical called ATP (adenosine tryphosphate). Without oxygen or nutrition, we can’t thrive. So we can’t just eat to live, we have to breathe as well. Breathing is the difference between being alive and dead — in more ways than one! But of course, the lungs can’t do their work unless they are already supplied with energy, which poses a “which came first: the chicken or the egg?” situation.
There are dozens of analogies to illustrate the biochemical process: cars running on gas or factories experiencing a brownout as well as descriptions of dysfunction effects: The Spoon Theory, bank account deposits vs. withdrawals or varying size of pie slices. Find one that speaks to you [LINKS].
Every person has some level of mitochondrial dysfunction – no one’s mitochondria function at 100%, all of the time. No Olympic athlete or person with a disability. Mitochondrial function is also a snapshot in time – changes in nutrition, lifestyle, and activity level all affect the mitochondrial processes. So can mito ever be really ruled out? Obviously, an Olympic athlete’s level of dysfunction isn’t a problem, but there can come a point, though, where the level of mitochondrial dysfunction impacts day-to-day living and your body systems’ health. Among “mito experts” there is no consensus of what constitutes “disease level” dysfunction: 35% if you score by modified Walker criteria as one of three major and two minor criteria; a six for “probable” or eight-plus for “definite” on the scale used by the Nijmegan Center for Mitochondrial Disorders; or 5% function if you listen to a researcher/specialist.
Genetics:
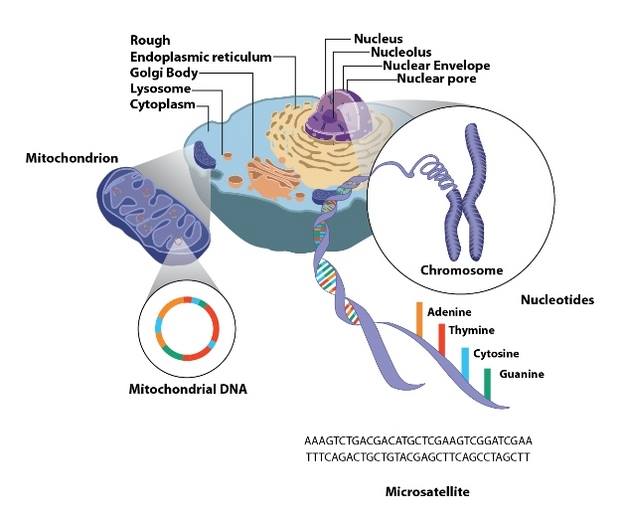
 The medical establishment’s contention has long been that mito is only inherited, 20% of cases through mtDNA and 80% with nDNA mutations driving their symptoms. Molecular genetics has changed dramatically with Whole Exome (or Genome) Sequencing, but even after close to 10 years, it is estimated that only 30% of patients get diagnosed using WES. What else could be influencing our mitochondrial health if not inherited genetics? (See my friend Kristi’s blog for some clues: www.baby(food)steps.com for M.I.T.O series.)
The medical establishment’s contention has long been that mito is only inherited, 20% of cases through mtDNA and 80% with nDNA mutations driving their symptoms. Molecular genetics has changed dramatically with Whole Exome (or Genome) Sequencing, but even after close to 10 years, it is estimated that only 30% of patients get diagnosed using WES. What else could be influencing our mitochondrial health if not inherited genetics? (See my friend Kristi’s blog for some clues: www.baby(food)steps.com for M.I.T.O series.)
In addition to WES, 23andme has also changed the landscape of genetics by analyzing DNA mutations from an ancestry perspective while also providing the raw data that can provide a picture of your health or unwellness, mutations that are not likely to be discovered with WES. Does the answer lie in personalized genetics? Maybe eventually. I’m not going to wait, though. My kids’ health now and future survival depend on choices we make daily.
Supplements:
Supplements are a topic with a mixed message from “mito experts” as well as local clinicians. The message has only become more confusing in 10 years. Some prescribe a customized cocktail, some suggest an over-the-counter list saying “take what you can afford,” and still others are influenced by Children’s Hospital of Philadelphia’s 2013 policy change. The mixed message evolved from a debate: if the cocktail was tailored to an individual, specific combinations could potentially improve symptoms. But the FDA doesn’t regulate supplements, which opens up a whole new can of “safety and efficacy” worms.
 Finally in 2009, a standard range of recommendations was collectively presented in “A Modern Approach to the Treatment of Mitochondrial Disease.” The collective recommendations from the top doctors was energizing. Basic mito cocktail (pharmacologic treatment) dosage recommendations were in writing, but the article also covered specific details under the topic headings of “Diet & Lifestyle,” “Immunizations,” and “Avoiding Toxins.” There is mention, too, of “Historical treatments now less commonly used,” interventional procedures such as IV fluids, anesthesia, and surgery. An all-encompassing document to guide “best practices,” this was followed up by a 2010/2013 version Mito 101 series from the advocacy group United Mitochondrial Disease Foundation, which contains much the same information. Eventually, combination supplement products appeared on the market. But still no studies have shown any supplements to be beneficial for specific phenotypes. Results will vary with each individual person.
Finally in 2009, a standard range of recommendations was collectively presented in “A Modern Approach to the Treatment of Mitochondrial Disease.” The collective recommendations from the top doctors was energizing. Basic mito cocktail (pharmacologic treatment) dosage recommendations were in writing, but the article also covered specific details under the topic headings of “Diet & Lifestyle,” “Immunizations,” and “Avoiding Toxins.” There is mention, too, of “Historical treatments now less commonly used,” interventional procedures such as IV fluids, anesthesia, and surgery. An all-encompassing document to guide “best practices,” this was followed up by a 2010/2013 version Mito 101 series from the advocacy group United Mitochondrial Disease Foundation, which contains much the same information. Eventually, combination supplement products appeared on the market. But still no studies have shown any supplements to be beneficial for specific phenotypes. Results will vary with each individual person.
Nutrition:
Every food can be broken down into a combination of fats, carbohydrates and proteins. Rarely, these processes don’t work, causing diseases collectively known as inborn errors of metabolism (IEMs). IEMs occur in the pathways leading to the mitochondria and directly contribute to the mitochondria’s function or dysfunction and affect whether the energy source is used or stored. IEMs include
- Fatty Acid oxidation disorders – can’t convert fats to energy, with the fats measured in chain length (short to medium to long to very long)
- Urea Cycle disorders – proteins can’t be broken down, treatment is limited dietary intake
- Carbohydrates – Glycosylation, glycogen storage disorders
The metabolic pathways that break food down all function to keep the cells churning out energy – at 600 cycles per second per cell. Every time you move a muscle, take a breath, have a thought, or digest food, your cells use the ATP they’ve just produced – no storage here! A mitochondrion creates that ATP molecule by a five-step process called oxidative phosphorylation (OXPHOS). Since there are five steps involved, it’s easy to see why there’s so much variability in mitochondrial dysfunction, all the way from asymptomatic to profoundly disabled.
Mitochondrial function and nutrition:
Mitochondrial-associated metabolic disorders: foundations, pathologies and recent progress:
Though clinical data investigating how diet influences mitochondrial function is currently quite limited, one well studied diet associated with influencing mitochondrial function is the ketogenic diet. The ketogenic diet is a diet rich in fat and low in carbohydrates, designed to stimulate ketone body biogenesis by replacing carbohydrates with fats as a primary energy source. As discussed below, many mitochondrial pathologies arise from defects in mitochondrial metabolism. In some cases of disease, mutations affect proteins required for glucose oxidation (also causing ETC inhibition), leading to the inability of glucose to serve as a primary carbon source.
Explaining the pathways of the energy sources, we can start to understand how nutrition is a key factor in maximizing energy production of mitochondria:
As discussed, energy sources provided by diet include carbohydrates and fats. Carbohydrates are broken down by the glycolysis pathway and TCA cycle to yield reduced NADH and FADH2, which are shuttled to the ETC for ATP production. Fats, on the other hand, are broken down byβ-oxidation (Beta-oxidation) to yield reduced NADH and FADH2, as well as acetyl-CoA, which will enter the TCA cycle. Additionally, in the liver and kidneys, ketone bodies are produced as a by-product of fatty acid β-oxidation. Ketone bodies are high-energy compounds which can be transported via the blood to other tissues where they are processed and enter the TCA cycle. Ketone bodies thus have the ability to serve as an alternative energy source for tissues in situations of impaired glucose oxidation.
So if you know your mutations, science and your medical team should be able to suggest a starting point for personalized nutrition. How many clinicians are doing just that — suggesting pharmacological solutions based on genetics? Are the WES laboratories making those suggestions now? They weren’t five years ago with our first round of results.
The author continues this novel idea, suggesting that the proper diet regime may alleviate symptoms:
As discussed below, nearly all mutations and defects associated with mitochondrial pathologies result in decreased activity of ETC complexes. Currently, in order to treat these diseases, researchers and clinicians must first identify the individual mutations and proteins involved in the pathology before attempting to design a therapy, which will only be useful for treating that specific form of disease. In contrast, diet appears to be able to directly influence mitochondrial metabolism, and therefore may have the ability to alleviate or suppress metabolic defects associated with mitochondrial pathologies. Therefore, by developing a diet regiment [sic] which increases metabolic function in cases of defects in mitochondrial function, one single diet plan may have the ability to treat a wide range of mitochondrial pathologies.
We are awaiting a re-examination by MEDomics, so we’ll see if their staff have anything new to recommend.
Munchausen Syndrome by Proxy (MSBP) & Child Protective Services (CPS):

Justina Pelletier before her CPS ordeal
When I first joined a mito parents’ group in 2005, there was a welcoming sense of support and guidance, which continued for 2-2 1/2 years.It was a small group of parents, about 100 or so, the majority with kids severely affected. I learned the most from a parent’s perspective those first years - pieces of our puzzle fit tightly, despite our doctors saying “they aren’t sick enough.” The group’s tone changed when several members were singled out for charges of medical abuse; doctor names couldn’t be used; newbie questions were viewed suspiciously instead instead of supportively; gossip about accused members escalated; many original members quit or went to rather less-known groups. Then Facebook scattered more people across the Internet, but also allowed more connections.
It all came to a head with Justina Pelletier, in 2013-2014. I vaguely remember the late-night post from a mom whose daughter was sick with GI/flu symptoms and asking for guidance about going in for IV fluids. Mom knew the potential for mito crashes, but without saying it in so many words was indicating that she was fearful of potential intervention by CPS due to previous conflicts. This fear permeates every mito discussion. Mom’s delayed response conveyed hidden meanings only mito families can understand. I knew my mito community would rally when the time came; her child needed to go in. Mom deleted the conversation, and I wondered what happened in the months following, eventually forgetting about it. Until word got out: The Pelletiers were taking on the Massachusetts CPS system and Boston Children’s Hospital. I have only experienced the level of sadness I had when I pieced the story together late one night one other time, when seven or more kids with mito died in one week — most of them unexpectedly — shocking the mito community to the core.
Future of mito:
I can only surmise that if the next 10 years are anything like the last 10, I do not want to be near a mito diagnosis. The families affected are not winning the battle, and the moneys raised by walkathons and cookbooks are funding what exactly? Genetic research and testing that apply to 20% of cases. WHY are the 80% ignored? Our mito docs are now expert witnesses in CPS cases across the country, instead of treating sick patients. They are closing clinics, sending patients scrambling to docs with six to eight-month wait lists. The patients are having to come up with out-of-pocket funds since these docs are not accepting insurance, or only patients within the state they’re located. Mito advocacy groups are aligning with Pharma, for EPI-743, Bendavia, RP103 and RTA 408 clinical trials. Where are the clinical studies on the causation factors — glyphosate, GMOs, vaccine ingredients, or whatever else might be suggested, like marshmallows? Is mito inherited as much as the studies claim? Or are genes being modified from toxic exposure? We may never find out since there’s no money in causation.
~ RogueZebra
Supplemental links on autism and mitochondrial disease:
The Journey From ASD to a Mitochondrial Disease Diagnosis
Neurotensin and Extra-Cellular Mitochondria: Potential Biomarkers and Novel Treatment Targets
For more by RogueZebra, click here.










This post ROCKS a absolutely perfect summary of the horrors of Mitochondrial Disease. Not only does it destroy one’s health, but it is destroying families, communities and Medicine in general. As long as the Mito Specialists continue to have tunnel vision with mutations the 80 percent who don’t have an in your face mutation that the Drs can rationalize is “inherited” we will be in big trouble. Some will die. Some will lose everything. Some will be ostracized by not only their community but by the Medical Community that diagnosed them in the first place. Until the Medical Community is will to admit that medications and other environmental toxins(vaccines, pesticides, herbicides, insecticides, industrial etc) are the likely cause of the 80 percent of Mito Patients who’s lives have been destroyed? A doomed population. The message is clear- Dollars count more then patients.
What I have been reading lately leads me to believe that gene expression is very fluid and changing all of the time in response to environmental factors. Its like a conversation, among sets of genes (vibrational, think light or music) and the environmental envelope. Apparently our genes change with the seasons, with different foods, in response to electrochemical and certainly to toxic chemicals. Dr Mae Wan Ho, a physicist who edits the Institute for Science in Society has written extensively about the cutting edge research.
Also, the way that ATP works in the medical text books has been found to be completely wrong. I´m trying to remember the details, think it was this video on structured water: https://www.youtube.com/watch?v=2edKFnVX5CE#t=2868
I think very few things concerning the mitochondria are genetic problems. The mitochondria is basically the motor, the energy producer. Gut dysbiosis, mineral deficiency, cipro (or any of the new antibiotics with a fluoride base) can sort of make it work at lower capacity.
I think a functional medicine approach is always best first. Do what you can to work with nature, pump up the nutrients in a balanced way with foods, and get the immune system functioning. We can´t afford testing anyways. We are drinking chia water, for the structured water effects. After 3 years of GAPS, and probiotics, fermented foods, and vitamin C, we´ve had quite a lot of symptoms go away.
Fluoroquinolone antibiotics - Cipro/ciprofloxacin, Levaquin/levofloxacin, Avelox/moxifloxacin, Floxin/ofloxacin, and a few others - deplete mitochondrial DNA and interfere with mitochondrial DNA and RNA replication and transcription, just like they do to bacteria. Here is an article about it - http://floxiehope.com/2015/02/24/study-finds-that-ciprofloxacin-depletes-mitochondrial-dna/
Also, the FDA said in a recent report:
“Ciprofloxacin has been found to affect mammalian topoisomerase II, especially in mitochondria. In vitro studies in drug-treated mammalian cells found that nalidixic acid and ciprofloxacin cause a loss of motichondrial DNA (mtDNA), resulting in a decrease of mitochondrial respiration and an arrest in cell growth. Further analysis found protein-linked double-stranded DNA breaks in the mtDNA from ciprofloxacin-treated cells, suggesting that ciprofloxacin was targeting topoisomerase II activity in the mitochondria.”
Please do not give your babies fluoroquinolones! They’re prescribed to children all the time for ear and eye infections. The damage done to both the microbiome and the mitochondria is horrifying.